
La primera benzodiacepina, el clordiacepóxido, fue sintetizada por casualidad en 1961. El insólito anillo de siete componentes se obtuvo como resultado de una reacción química que no funcionó como cabía esperar en los laboratorios de Hoffman-La Roche. Su inesperada actividad farmacológica fue identificada en un procedimiento rutinario de cribado y muy pronto las benzodiacepinas se convirtieron en los fármacos más ampliamente prescritos de la farmacopea.
La estructura química básica de las benzodiacepinas consiste en un anillo de siete elementos unido a un anillo aromático, con cuatro posibles sustituciones que pueden ser modificadas sin pérdida de actividad.
Se han sintetizado y probado miles de compuestos y alrededor de 20 se hallan disponibles para su uso clínico, siendo algunos de los más importantes los que figuran en la relación de la tabla 43.1.

Básicamente, son similares en cuanto a acciones farmacológicas, aunque se ha referido cierto grado de selectividad. Por ejemplo, algunos de ellos, como el clonacepam, muestran actividad anticonvulsionante, con efectos sedantes menos marcados. Desde el punto de vista clínico, las diferencias en cuanto al comportamiento farmacocinético entre las distintas benzodiacepinas son más importantes que las diferencias en cuanto a su perfil de actividad. Se ha descubierto que fármacos de estructura similar tienen una acción específica antagonista de los efectos de las benzodiacepinas, como es el caso del flumacenilo
.
El término «benzodiacepina» hace referencia a una estructura química definida. Fármacos como el zolpidem y la zopiclona tienen una estructura química diferente y, por consiguiente, no son benzodiacepinas. Sin embargo, dado que se unen a los mismos sitios, denominados a menudo «receptores de benzodiacepinas», se habla de ellos al mismo tiempo que de las benzodiacepinas.
MECANISMO DE ACCIÓN
Las benzodiacepinas poseen una acción selectiva sobre los receptores GABAA, que actúan en el sistema nervioso central como mediadores en la transmisión sináptica inhibidora.
Las benzodiacepinas potencian la respuesta al GABA al facilitar la apertura de los canales de cloruro activados por dicho transmisor (fig. 43.4).
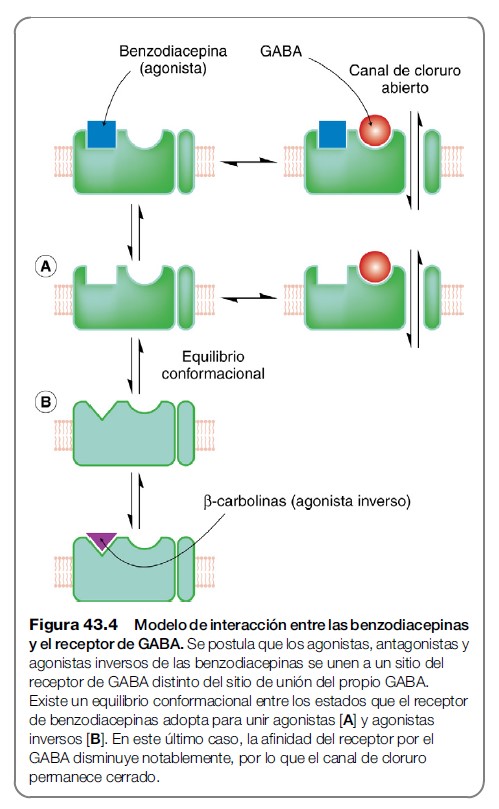
Se unen de manera específica a un sitio regulador del receptor diferente de los sitios de unión del GABA , y actúan de manera alostérica incrementando la afinidad del GABA por el receptor. Los registros de canal único muestran un incremento de la frecuencia de apertura del canal para una concentración dada de GABA, pero no revelan cambios de conductancia ni del tiempo medio de apertura, lo que concuerda con un efecto sobre la unión del GABA más que sobre el mecanismo de apertura del canal. Las benzodiacepinas no afectan a los receptores para otros aminoácidos, como la glicina o el glutamato (fig. 43.1).

El receptor GABAA es un canal iónico activado por ligando que consiste en una estructura pentamérica de diferentes subunidades, siendo las principales a, b y g . En realidad, el receptor GABAA debe considerarse como una familia de receptores, pues existen seis subtipos diferentes de subunidades a, tres subtipos de b y tres subtipos de g. Aunque el número potencial de combinaciones es, por consiguiente, elevado, en el cerebro adulto predominan algunas de ellas . Las distintas combinaciones tienen lugar en diferentes partes del cerebro, tienen distintas funciones fisiológicas y muestran sutiles diferencias en cuanto a sus propiedades farmacológicas .
Las benzodiacepinas se unen en la interfase entre las subunidades a y g, aunque solo a los receptores que contienen las subunidades g2 y a1, a2, a3 o a5. Se han utilizado dos enfoques genéticos para estudiar el papel de las diferentes subunidades en los distintos efectos conductuales de las benzodiacepinas: el bloqueo genético y los mutantes con pérdida de función (v. Whiting, 2003; Reynolds, 2008). El enfoque del mutante de pérdida de función tiene la ventaja, sobre el bloqueo de la subunidad, de que reduce la probabilidad de cambios compensatorios en la expresión de otras subunidades. La mutación de un aminoácido aislado (histidina 101 o su equivalente) en la subunidad a elimina la unión de la benzodiacepina. El análisis conductual de varios ratones mutantes indica que los receptores que contienen a1 median en el efecto sedante de las benzodiacepinas, pero no en el ansiolítico, mientras que los receptores que contienen a2 y a3 median en el efecto ansiolítico.
El paso siguiente ha sido, evidentemente, probar y desarrollar fármacos selectivos para las distintas subunidades (Reynolds, 2008; Christmas et al., 2008). Por desgracia, dicha tarea ha resultado difícil debido a la similitud estructural en el sitio de unión de las benzodiacepinas entre las diferentes subunidades a. Lo que sí ha sido posible es el desarrollo de fármacos que, aun teniendo escasa selectividad de unión a una subunidad, muestran distintos niveles de eficacia agonista sobre los receptores que contienen diferentes subunidades. La eficacia selectiva en los receptores que contienen a2 y a3 puede producir fármacos con efecto ansiolítico y sin los efectos adversos de sedación y amnesia. Tales compuestos han sido desarrollados (p. ej., MK-0343, TPA023), aunque actualmente solo se dispone de datos limitados sobre su eficacia en seres humanos. El pagoclone, que según se ha referido es agonista de a3 y agonista parcial de a1, a2 y a5, posee escasa o nula acción sedante o amnésica y se halla en desarrollo para el tratamiento de los trastornos de angustia y la tartamudez.
Se sabe que en muchos tejidos existen sitios periféricos de unión de las benzodiacepinas no asociados a receptores de GABA. Se localizan fundamentalmente en las membranas mitocondriales.
Para más información sobre su estructura y funciones, véase Veenman y Gavish (2006).






