CHILE : http://www.ispch.cl/farmacovigilancia
Para demostrar la eficacia, la FDA requiere la realización de “investigaciones adecuadas y bien controladas”, por lo general interpretadas por medio de dos estudios clínicos que dupliquen los resultados y que habitualmente, aunque no siempre, son realizados con asignación al azar, doble ciego y con grupo testigo que recibió placebo.
¿Es el placebo un mecanismo de control apropiado?
La declaración de Helsinki de la World Medical Association (2000) no recomienda el uso de grupos testigos con placebo cuando se dispone de un tratamiento alternativo con fines de comparación.
¿Qué debe cuantificarse en los estudios clínicos?
En un estudio simple, prospectivo, se mide un parámetro fácilmente cuantificable (un criterio de valoración secundario) que sea capaz de predecir resultados clínicos relevantes y se compara con los grupos que recibieron el fármaco de referencia o el placebo. Ejemplos de puntos de valoración secundarios incluyen la concentración de colesterol LDH como factor pronóstico de infarto miocárdico, la densidad mineral ósea para el pronóstico de fracturas, o la hemoglobina A1c como pronóstico de la diabetes mellitus.
Los estudios más estrictos requieren la demostración de una reducción en la incidencia de infarto miocárdico en pacientes que reciben un inhibidor de la HMG-CoA reductasa (estatinas) u otro fármaco que reduzca las concentraciones de LDL o bien, reducción en la incidencia de fracturas al comparar el fármaco estudiado con aquellos pacientes que reciben bisfosfonatos. El uso de criterios de valoración reduce de manera significativa los costos y el tiempo necesario para completar los estudios clínicos, pero existen muchos factores atenuantes, lo que incluye la importancia del punto de valoración para la enfermedad que se intenta tratar.
Algunas de las dificultades se ilustran bien con experiencias recientes con ezetimiba, un fármaco que inhibe la absorción de colesterol del tubo digestivo y reduce las concentraciones de LDL en plasma, en especial cuando se utiliza en combinación con una estatina. Se supuso que la reducción de LDL era un punto de valoración apropiado para conocer la eficacia de ezetimiba en la reducción de infarto miocárdico y apoplejía como consecuencia de acumulación del colesterol en células espinosas que se encontraban por debajo del endotelio vascular.
De manera sorprendente, el estudio clínico ENHANCE demostró que la combinación de ezetimiba con una estatina no disminuía el grosor de las capas íntima y media de las arterias carótidas (una medición más directa de la acumulación de colesterol subendotelial) en comparación con el grupo que recibió solamente estatinas, pese al hecho que la combinación farmacológica redujo las concentraciones de LDL de manera muy sustancial en comparación con cualquiera de los fármacos solos (Kastelein et al., 2008).
Los críticos del estudio ENHANCE argumentaron que los pacientes en el estudio tenían hipercolesterolemia familiar, que habían recibido estatinas por años y que no tenían engrosamiento de las arterias carótidas al inicio del estudio.
¿Es necesario regresar a las mediciones de los dos criterios de valoración (p. ej., infarto miocárdico) antes de la aprobación de fármacos que reducen el colesterol por mecanismos novedosos?
información ejemplo actualizada :
Combination Therapy of Rosuvastatin and Ezetimibe in Patients with High Cardiovascular Risk.


Alguien debe pagar los costos relacionados con la realización de estudios amplios y costosos. Se está llevando a cabo un estudio de ese tipo (IMPROVE-IT) y en pocos años se conocerán los resultados de miles de pacientes incluidos en el estudio.
El fármaco torcetrapib proporciona un ejemplo relacionado en la misma área terapéutica; incrementa la concentración de colesterol HDL (“colesterol bueno”) y las concentraciones elevadas de HDL tienen asociación estadística con disminución en la incidencia de infarto miocárdico (un criterio de valoración).
De manera sorprendente, la administración clínica de torcetrapib ocasionó incremento significativo en la tasa de mortalidad por eventos cardiovasculares, terminando una vía de desarrollo de 15 años de duración con una inversión de 8000 millones de dólares (un análisis de sistemas biológicos por ordenador pueden explicar el fracaso, véase Xie et al., 2009). En este caso, la aprobación de un fármaco basado en un criterio de valoración secundario hubiera sido un error (Cutler, 2007).
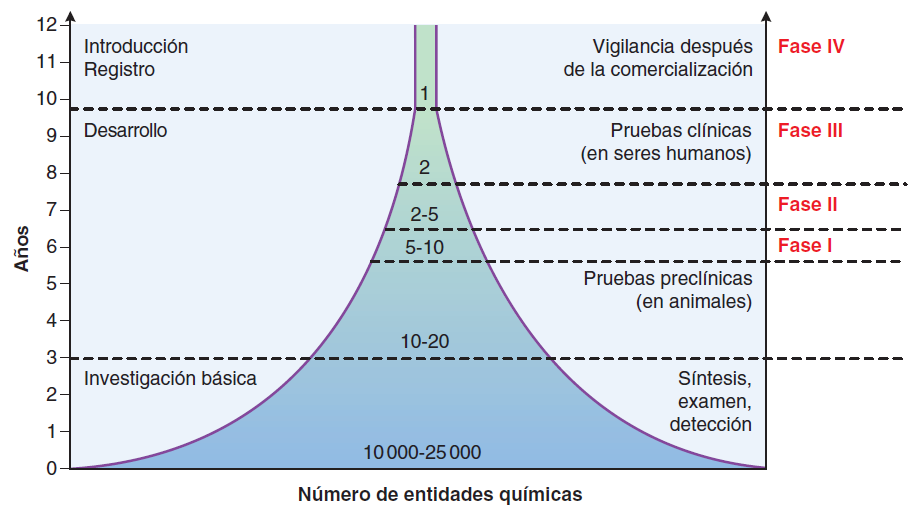
El concepto de seguridad farmacológica es quizá aún más complejo (Institute of Medicine, 2007). Ningún fármaco es por completo seguro; todos los medicamentos producen efectos indeseables en al menos algunas personas con la misma dosis. Muchos efectos graves e indeseables ocurren con baja frecuencia, quizá sólo una vez en varios miles de pacientes, de forma que permanecerían sin detección en poblaciones relativamente pequeñas (unos cuantos miles) en los estudios clínicos de fase III.
Para detectar y verificar tales eventos relacionados con los fármacos, sería necesaria la administración de un fármaco a decenas o cientos de miles de personas durante los estudios clínicos, lo que añadiría enormes costos y tiempo para el desarrollo de fármacos y el retraso de posibles beneficios terapéuticos. En general, el verdadero espectro e incidencia de efectos indeseables se conoce sólo después de la comercialización del fármaco a un mercado más amplio y el uso por grandes cantidades de personas (vigilancia después de la comercialización, estudios de fase IV). Los costos del desarrollo de fármacos y por tanto de los precios de los medicamentos, se podrían reducir de manera sustancial si el público estuviera dispuesto a aceptar mayores riesgos. Esto requeriría un cambio en la forma en que se piensa con respecto a la responsabilidad de las compañías farmacéuticas por daños producidos por efectos indeseables de un medicamento que no se detectaron en los estudios clínicos que parecieron adecuados para la FDA.
Mientras que el concepto es obvio, muchos pierden de vista el hecho de que los efectos indeseables extremadamente graves de un medicamento, lo que incluye la muerte, podrían parecer aceptables si su efecto terapéutico es suficientemente útil y singular. Tales dilemas se vuelven tema de gran debate. La suficiencia del efecto terapéutico en presencia de un efecto indeseable de un fármaco puede ser bastante subjetivo. Lo que beneficia a una persona puede ser veneno para otro. Se han realizado grandes esfuerzos para cuantificar la razón de riesgo/beneficio, pero las respuestas con frecuencia no son simples.
Existen varias estrategias para detectar reacciones adversas después de la comercialización de un fármaco, pero continúa el debate con respecto al método más eficiente y eficaz. Los métodos formales para la estimación de la magnitud de una respuesta adversa a medicamentos incluye estudios de vigilancia de pacientes que recibieron un fármaco en particular; el estudio de casos y testigos, donde la frecuencia del uso del fármaco en casos de respuestas adversas se compara con un grupo de testigos y metaanálisis de estudios realizados antes y después de la comercialización.
Por los inconvenientes de este tipo de estudios para detectar acontecimientos relativamente poco comunes, pueden utilizarse métodos adicionales. Los informes espontáneos de reacciones adversas han demostrado ser una manera eficaz para generar signos tempranos de que un fármaco podría causar eventos adversos (Aagard y Hansen, 2009). Es la única forma práctica de detectar eventos poco comunes, eventos que ocurren después del uso prolongado de fármacos, aquellos que aparecen de manera tardía y muchas interacciones farmacológicas. En fecha reciente se han realizado esfuerzos considerables para mejorar los sistemas de reporte en Estados Unidos, con un procedimiento conocido como MedWatch (Brewer y Colditz, 1999; Kessler et al., 1993).
Los sistemas de reporte voluntario en Estados Unidos no son tan potentes como los sistemas de información obligatorio de otros países. Un número considerable de médicos no está consciente de que la FDA tiene un sistema de reporte de reacciones farmacológicas adversas, incluso aunque el sistema se ha publicado de manera repetida en las principales publicaciones médicas (Trontell, 2004). Relativamente pocos médicos reportan en realidad respuestas farmacológicas adversas; aquéllas que se reciben con frecuencia están incompletas o son de mala calidad, de forma que los datos no se consideran fiables (Fontarosa et al., 2004).
Los reportes espontáneos más importantes son aquellos que describen reacciones graves. Los reportes sobre fármacos de comercialización reciente (en los primeros cinco años después de la introducción del medicamento) son los más significativos, aunque los médicos podrían no tener la capacidad de atribuir una función causal a un medicamento en particular.
Este sistema proporciona señales de alarma de efectos adversos inesperados y que pueden investigarse por medio de técnicas más formales. Sin embargo, el sistema también sirve para vigilar cambios en la naturaleza o frecuencia de reacciones medicamentosas adversas por envejecimiento de la población, cambios en la enfermedad misma o la introducción de nuevos tratamientos concomitantes.
La fuente primaria de los reportes son los médicos alertas y responsables; otras fuentes potencialmente útiles son farmacéuticos, enfermeras y estudiantes en estas disciplinas. Además, los comités de farmacia y de terapéutica en los hospitales y los comités para aseguramiento de la calidad con frecuencia se encargan de vigilar las reacciones medicamentosas adversas en pacientes hospitalizados y los reportes de estos comités deben ser enviados a la FDA.
Las formas para reporte pueden obtenerse 24 horas al día, siete días a la semana al llamar en Estados Unidos al número telefónico 800-FDA-1088; también pueden reportarse de manera directa utilizando la Internet (www.fda.gov/medwatch). Asimismo, los profesionales de la salud pueden establecer contacto con el fabricante farmacéutico, quien está legalmente obligado a reportarlo a la FDA. Con este sistema de reporte fácil, el médico puede actuar como centinela en la detección de reacciones adversas inesperadas a fármacos.
CHILE : http://www.ispch.cl/farmacovigilancia
En Chile la Farmacovigilancia esta dada por el Centro Nacional de Información de Medicamentos y Farmacovigilancia, del Instituto de Salud Pública de Chile. Está encargado de entregar información objetiva y evaluada sobre medicamentos, tanto a los profesionales de la salud del país, como al público en general. Además, tiene a su cargo el Programa Nacional de Farmacovigilancia.
Farmacovigilancia: Ciencia y conjunto de actividades relacionadas con la detección, evaluación, comprensión y prevención de los efectos adversos asociados al uso de los medicamentos.
Dentro del marco del Programa Nacional de Farmacovigilancia, CENIMEF recibe notificaciones de sospechas de reacciones adversas a medicamentos (RAM), reportadas por los profesionales de la salud del país, contribuyendo de esta manera, a la evaluación permanente y sistemática del perfil de seguridad de los medicamentos utilizados por la población nacional. Estos reportes son evaluados y procesados por un comité de expertos.
Funciones principales:
- Entregar información de medicamentos objetiva y evaluada, principalmente a los profesionales de la salud del país y al público en general.
- Monitorizar y evaluar sistemáticamente las reacciones adversas asociadas al uso de los medicamentos (RAM).
- Monitorizar y evaluar sistemáticamente la información de seguridad de medicamentos publicada en las principales Agencias Internacionales de Medicamentos.
Reacción Adversa a Medicamentos (RAM): Reacción nociva y no intencionada que se produce a dosis utilizadas normalmente en el hombre, con fines de curación, atenuación, tratamiento, prevención o diagnóstico de las enfermedades o sus síntomas, o para modificar sistemas fisiológicos
RAM Grave: son aquellas RAM que pueden producir alguna de las siguientes condiciones:
- Muerte o que pone en riesgo la vida.
- Hospitalización o prolongación de ésta.
- Incapacidad permanente o transitoria.