Los estudios clínicos (como se llevan a cabo para los fármacos) son investigaciones en seres humanos dirigidas a obtener información con respecto a las propiedades farmacocinéticas y farmacodinámicas de un fármaco potencial.
Dependiendo de la naturaleza y fase del estudio, puede estar diseñada para valorar la seguridad de un fármaco, su eficacia para el tratamiento o prevención de trastornos específicos y su tolerabilidad y efectos secundarios. Debe demostrarse que la eficacia tiene un margen adecuado de seguridad establecida para que se apruebe la comercialización del medicamento en Estados Unidos. Los U.S. National Institutes of Health hacen énfasis en siete requerimientos éticos que deben satisfacerse antes de iniciar un estudio clínico. Éstos incluyen utilidad social, validez científica, justicia y la selección objetiva de los sujetos, consentimiento informado, razón favorable de riesgo/beneficios, aprobación y vigilancia por un comité revisor independiente (IRB, independent review board) y respeto por los seres humanos.
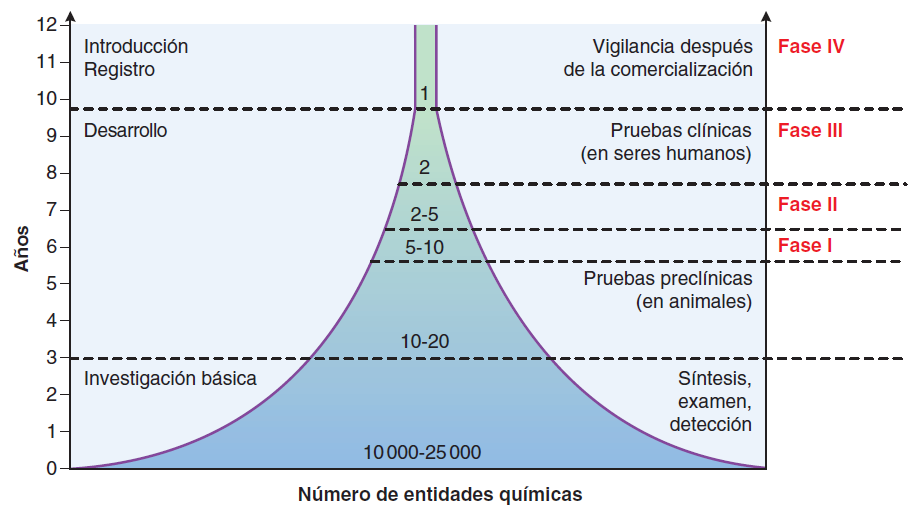
Los estudios clínicos regulados por la FDA por lo común se llevan a cabo en cuatro fases. Las primeras tres fases se diseñaron para establecer la seguridad y eficacia, mientras que la fase IV, que corresponde a los estudios realizados después de la comercialización, proporcionan información adicional con respecto a nuevas indicaciones, riesgos y dosis y esquemas óptimos. en el siguiente cuadro se resumen características importantes de los estudios clínicos de cada fase, en especial el agotamiento de cada etapa sucesiva sobre un proceso relativamente largo y costoso.


Cuando se han completado los estudios de fase III, el patrocinador (por lo común una compañía farmacéutica) solicita la aprobación ante la FDA para la comercialización del fármaco; esta solicitud se conoce como solicitud de un nuevo fármaco (NDA, New Drug Application) o como solicitud de licencia biológica (BLA, Biologics License Application).
Dichas solicitudes contienen información amplia, lo que incluye reportes de casos de cientos o miles de individuos que han recibido el fármaco durante las pruebas de fase III. Las solicitudes son realizadas por grupos de especialistas y la FDA podría solicitar el auxilio de grupos de expertos externos para la valoración de casos complejos. El uso de tales comités revisores externos incrementa en gran medida el talento disponible para ayudar en la toma de decisiones difíciles e importantes.
Bajo las previsiones de la Prescription Drug Use Fee Act (PDUFA; promulgada en 1992 y revisada en el año 2007), las compañías farmacéuticas proporcionan una parte significativa del presupuesto de la FDA a través del pago de honorarios, en un esfuerzo legislativo para hacer más expedito el proceso de aprobación de nuevos fármacos. PDUFA también amplió el programa de seguridad farmacológica de la FDA e incrementó los recursos para la publicación de anuncios televisivos de fármacos. Con el incremento en el personal de la FDA, se ha acortado el tiempo necesario para la revisión; no obstante, el proceso es aún prolongado.
Se considera como estándar un tiempo de revisión de un año y de seis meses si el posible fármaco ha logrado un estado de prioridad por su importancia al satisfacer una necesidad no satisfecha. Por desgracia, casi nunca se cumplen estos objetivos.
Antes de que un fármaco se apruebe para comercialización, la compañía y la FDA deben estar de acuerdo en el contenido del “prospecto de envase”, la información oficial para prescripción. Dicho prospecto describe las indicaciones aprobadas para el fármaco e información farmacológica clínica lo que incluye dosis, reacciones adversas y precauciones especiales (con mucha frecuencia ublicadas en un recuadro negro). Los materiales promocionales utilizados por las compañías farmacéuticas no deben desviarse de la información contenida en el prospecto de envase.
Es de gran importancia que el médico no está limitado a la información contenida en el prospecto de envase; un médico en Estados Unidos y en Chile puede prescribir de manera legal un fármaco para cualquier propósito que considere razonable. Sin embargo, las compañías aseguradoras por lo general no reembolsan los gastos por fármacos utilizados con indicaciones no reconocidas a menos que el nuevo uso se apoye por al menos en uno de varios compendios como la farmacopea estadounidense.
Además, un médico está expuesto a demandas si aparecen efectos indeseables por el uso de un fármaco con indicación no aprobada.