Calculo de Energias VDW, y Elec.
Es necesario poseer .psf .pdb .dcd. y especificar los 2 componentes a medir.
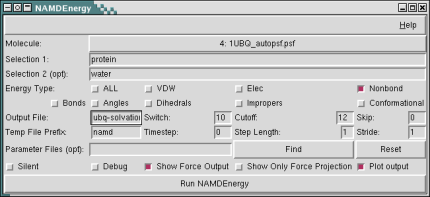
Basic Usage
NAMDEnergy operates on either one or two selections; if only one selection is chosen, then internal energies for that selection will be calculated, whereas if two selections are chosen, only interaction energies between those selections will be calculated. Selections are given using the standard VMD atom selection language.
In addition to one or two atom selections, the user must also choose one or more energy types to be calculated. Choices are: bonds, angles, dihedrals, impropers, vdW energy, electrostatic energy, conformational energy (bonds, angles, dihedrals, and impropers), nonbond energy (vdW and electrostatic energy), or all (all other energies). Energies will either be printed to the console or an optionally specified output file. Note that only nonbond energy types are available for interactions between two selections. Note that all energy output is given in kcal/mol, and all force outputs in kcal/(mol Å). Multiple parameter files may be given by specifying the -par switch multiple times.
PME and Periodic Cells
Most trajectories which were performed with periodic boundary conditions will include periodic cell information for each step in the DCD file. However, for proper initialization of a periodic system NAMD must also be fed an initial guess for the periodic cell, which will then be replaced by the data from the trajectory as NAMDenergy runs. This means that if you are running NAMDEnergy on a DCD produced from a periodic run, you should set the “XSC File” option to point to an xsc file from your trajectory. If you don’t have one available, click “Generate” and a GUI will allow you to create an appropriate one. If and only if you have a periodic system specified, you can also enable PME by checking the PME box; an appropriate mesh density will be automatically specified.
Text Interface
All options in NAMDEnergy are immediately visible in the GUI, but a text interface is also provided for scripting purposes. In the text interface, energy types are indicated by the switches -bond -angl -dihe -impr -conf -vdw -elec -nonb -all; one or two atom selections should also be given after a -sel switch to define the atom selections to run energy calculations on. The argument for the -sel switches is a variable containing either one or two atom selections; if a single selection is given (i.e., -sel $sel1), then energies for that selection are calculated. If two selections are given (-sel $sel1 $sel2), only interactions between the two selections will be calculated. When using two selections take care that the second atom select does not include any atoms from the first. All other optional parameters use switches defined in the following section. The text version can be called using the command namdenergy once the namdEnergy package has been loaded. It will return a list of lists, where the first element is a list of the energy headers being output, and each subsequent element is a list of the energy outputs.




