

Cuando se comenzaron a usar en seres humanos, los antimicrobianos se consideraron curas milagrosas. Sin embargo, poco después del descubrimiento de la penicilina se advirtió que aparecía en forma rápida resistencia, lo que ponía fin al milagro.
Esta situación grave persiste con cada antimicrobiano nuevo y amenaza con terminar con la época de los antimicrobianos.
En la actualidad, cualquier clase importante de antibióticos se acompaña de la aparición de resistencia notable.
Dos factores importantes se vinculan con este fenómeno:
Evolución y prácticas clínicas y ambientales.
Una especie sometida a presiones químicas o de otro tipo que amenazan con su extinción, suele desarrollar mecanismos para sobrevivir bajo tal tensión excesiva.
Los patógenos evolucionarán para presentar resistencia a la “guerra química” a la que están sometidos.
Esta evolución se facilita con prácticas terapéuticas inadecuadas realizadas por el personal de atención de la salud, así como por el uso indiscriminado de antibióticos con fines agrícolas o de cría de animales.
Las prácticas clínicas inadecuadas que no incorporan las propiedades farmacológicas de los antimicrobianos aceleran el surgimiento y la evolución de la resistencia a fármacos.
La resistencia a antimicrobianos puede surgir en una o más de las etapas de los procesos por los que el fármaco llega y se combina con el sitio en que actúa. De este modo,
la resistencia puede surgir a causa de:
- Resistencia por la menor penetración del fármaco en el interior del patógeno.
- Resistencia causada por expulsión del fármaco.
- Resistencia por destrucción del antibiótico.
- Resistencia surgida por la menor afinidad del fármaco a estructuras
blanco alteradas. - Alteración de las proteínas en que actúa un fármaco
- Creación de otras vías distintas a las inhibidas con el antibiótico.Los mecanismos por los cuales surge esta resistencia incluyen la adquisición de elementos genéticos que codifican el mecanismo de resistencia, mutaciones que aparecen con la presión ejercida por los antibióticos e inducción constitutiva.
Resistencia por la menor penetración del fármaco en el interior del patógeno.
La membrana externa de las bacterias gram negativas es una barrera permeable e impide que grandes moléculas polares penetren en el microorganismo.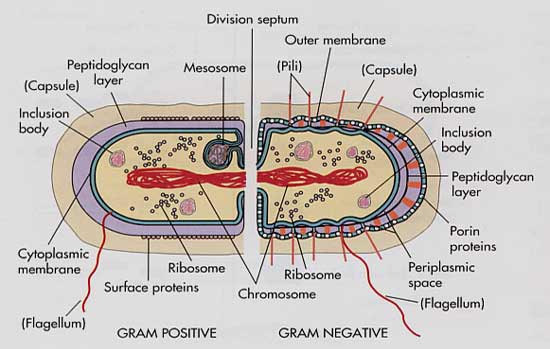
Penetran en él las pequeñas moléculas polares, como las de muchos antibióticos, a través de conductos proteínicos llamados porinas. La ausencia de un conducto de porina, su mutación o la desaparición de un conducto preferido desaceleran el ritmo de penetración del fármaco en un microorganismo o impiden que penetre, lo que disminuye en efecto la concentración del fármaco en el sitio en que debe actuar.
Si este sitio está en el interior de la célula y el fármaco necesita transporte activo para cruzar la membrana celular, una mutación o cambio fenotípico que anule o retrase el mecanismo de transporte conferirá resistencia.
Por ejemplo, la infección por rypanosoma brucei es tratada con suramina y pentamidina en sus fases iniciales, pero con melarsoprol y eflornitina cuando se produjo ya la enfermedad del SNC (enfermedad del sueño). El melarsoprol es captado en forma activa por la proteína transportadora P2 del tripanosoma.
Si el parásito no cuenta con dicha transportadora P2 o tiene una forma mutante, surgen resistencia al melarsoprol y resistencia cruzada a la pentamidina por una menor captación del transportador (Ouellette, 2001).
Suramina es el nombre de un medicamento antihelmíntico, desarrollado en Alemania en 1916 y aún en el mercado, indicado para el tratamiento de la tripanosomiasis africana.
Resistencia causada por expulsión del fármaco.
Los microorganismos a veces expresan en exceso bombas de expulsión y así
expelen antibióticos a los que, en otras circunstancias, serían susceptibles.
Se conocen cinco sistemas de bombas de expulsión que son importantes para los antimicrobianos:
- el elemento encargado de expulsión de compuestos tóxicos y múltiples fármacos (MATE)
- transportadores mayores de la superfamilia de facilitadores (MFS)
- el sistema pequeño de resistencia a múltiples fármacos (SMR)
- los exportadores de la división de modulación de resistencia (RND)
- los transportadores del casete de unión a ATP (ABC)
Las bombas de expulsión constituyen un mecanismo notable de resistencia de parásitos, bacterias y hongos. Una de las consecuencias trágicas de la aparición de resistencia ha sido la de Plasmodium falciparum. La resistencia a casi todos los antipalúdicos
y en particular a cloroquina, quinina, mefloquina, halofantrina, lumefantrina y la combinación de artemeter-lumefantrina es mediada por un transportador ABC que codifica el gen 1 de resistencia a múltiples fármacos, propio de Plasmodium falciparum
(Pfmdr1) (Happi et al., 2009). Las mutaciones puntuales en dicho gen originaron resistencia farmacológica e ineficacia de la quimioterapia.

Plasmodium falciparum. Es un protozoo parásito, una de las especies del género Plasmodium que causa malaria en humanos. Es transmitida por mosquitos Anopheles.Antipalúdico. Los fármacos antipalúdicos o antimaláricos, son medicamentos diseñados para prevenir o curar la malaria.
La expulsión del fármaco a veces actúa en tándem con la resistencia cromosómica, como se observa en Streptococcus pneumoniae y, tal vez, en Mycobacterium tuberculosis.
En estas situaciones surge pronto la inducción de las bombas de expulsión y aumentan muy poco la MIC. Sin embargo, dicho incremento de MIC basta para que surja más replicación microbiana y aumente la frecuencia de mutación, lo que permite que aparezca resistencia por medio de mutaciones cromosómicas más sólidas (Gumbo et
al., 2007b; Jumbe et al., 2006).
Resistencia por destrucción del antibiótico.
La inactivación del fármaco es un mecanismo frecuente de resistencia a él.
La resistencia bacteriana a aminoglucósidos y antibióticos lactámicos β en general depende de la producción de una enzima modificadora de aminoglucósido o de una lactamasa β, respectivamente.
Resistencia surgida por la menor afinidad del fármaco a estructuras
blanco alteradas.
Una consecuencia frecuente de mutaciones puntuales únicas o múltiples es el cambio de la composición de aminoácidos y la conformación de la proteína blanco. Este cambio
hará que disminuya la afinidad del fármaco por su sitio de acción o de un profármaco por la enzima que lo transforma en fármaco activo.
Dichas alteraciones pueden deberse a la mutación del sitio de destino natural (p. ej., la resistencia a la fluoroquinolona), la modificación del sitio de acción (p. ej., el tipo de protección ribosómica de la resistencia a los macrólidos y las tetraciclinas) o la adquisición de una forma resistente de la célula blanco susceptible y nativa (p. ej., la resistencia del estafilococo a la meticilina causada por la generación de una proteína de unión con poca afinidad por la penicilina) (Hooper, 2002; Lim y Strynadka, 2002; Nakajima, 1999).
En forma similar, en la resistencia de VIH se observan mutaciones asociadas a la reducción de la afinidad en el caso de los inhibidores de proteasa e integrasa, inhibidores de fusión y de transcriptasa inversa no nucleosídicos (Nijhuis et al., 2009).
De igual modo, los benzimidazoles se utilizan contra miles de helmintos y protozoos y actúan al unirse a la tubulina del parásito; mutaciones puntuales en el gen de la tubulina β originan modificación de dicha proteína y resistencia al fármaco (Ouellette, 2001).
Incorporación del fármaco.

Surge una situación poco frecuente cuando un microorganismo no sólo crea resistencia a un antimicrobiano, sino que después lo necesita para proliferar.
Los enterococos, que presentan con facilidad resistencia a la vancomicina, después de exposición prolongada al antibiótico terminan por desarrollar cepas que necesitan vancomicina.
Resistencia por intensificación de la expulsión del fármaco incorporado.
Los inhibidores nucleosídicos de la transcriptasa inversa como la zidovudina son análogos de 2′-desoxirribonucleósido que son convertidos en su forma de 5′-trifosfato y compiten con los nucleótidos naturales. Estos fármacos son incorporados en la cadena
de DNA del virus y la terminan. Cuando la resistencia surge por mutaciones en diversos puntos del gen de la transcriptasa inversa, se intensifica la expulsión fosforolítica del análogo nucleosídico incorporado, cuya función es terminar la cadena (Arion et al., 1998).
Zidovudina, Azidotimidina o AZT fue el primer medicamento antirretroviral (ARV), aprobado en 1987 como un medicamento indicado para personas infectadas con el VIH por su efecto retardador de la extensión de la infección por VIH, aunque no representa una cura y no garantiza la disminución de la cantidad de enfermedades relacionadas con la infección por VIH. La zidovudina reduce la transmisión del VIH a otras personas. Es comercializado bajo el nombre de Retrovir y Retrovis, y es un ingrediente en el Combivir, Epzicom y Trizivir. Es un análogo de la timidina.
La eritropoyetina se usa para contrarrestar la anemia producida por la zidovutina.
Heterorresistencia y cuasiespecies virales.
Se dice que hay heteroresistencia cuando un subgrupo de la población microbiana total es resistente, a pesar de que la población total se considere susceptible en las pruebas (Falagas et al., 2008; Rinder, 2001). En cierta forma, no debe causar sorpresa dado que las mutaciones cromosómicas siguen un esquema estocástico y hay un índice de mutación basal para cada gen. En consecuencia, se espera que una subclona que muestra alteraciones en genes asociados a la resistencia farmacológica refleje los índices de mutación normal, y que tienen lugar entre los 10−6 y 10−5 colonias. En las bacterias se ha descrito la heterorresistencia en especial contra la vancomicina en el caso de S. aureus , contra el mismo antibiótico en el caso de Enterococcus faecium, contra la colistina en Acinetobacter baumannii-calcoaceticus; contra la rifampicina, la isoniazida y la estreptomicina en el caso de M. tuberculosis, y contra la penicilina en el caso de S. pneumoniae (Falagas et al., 2008; Rinder, 2001). En individuos con estafilococos y M.
tuberculosis heterorresistentes se ha señalado un número cada vez mayor de ineficacias terapéuticas y muerte (Falagas et al., 2008; Hofmann-Thiel et al., 2009).
En el caso de los hongos, se ha descrito heterorresistencia que condujo a ineficacia clínica en el caso del fluconazol en el ataque por Cryptococcus neoformans y Candida albicans (Marr et al., 2001; Mondon et al., 1999).
La replicación viral está más predispuesta a errores que la replicación de bacterias y hongos. La evolución del virus bajo presiones medicamentosas e inmunitarias surge con facilidad relativa y suele originar variantes o cuasiespecies que pueden contener subpoblaciones farmacorresistentes. No se conoce a lo anterior como heterorresistencia, pero el principio es el mismo que el descrito para bacterias y hongos. Se puede considerar que un virus es susceptible a un fármaco porque en los estudios fenotípicos o genotípicos se detecta falta de resistencia, cuando existe una subpoblación resistente por debajo del límite de detección del método de cuantificación. Estas cuasiespecies minoritarias que son resistentes a los antirretrovirales se han vinculado con ineficacia del tratamiento antirretroviral (Metzner et al., 2009).





