
Dinamica molecular
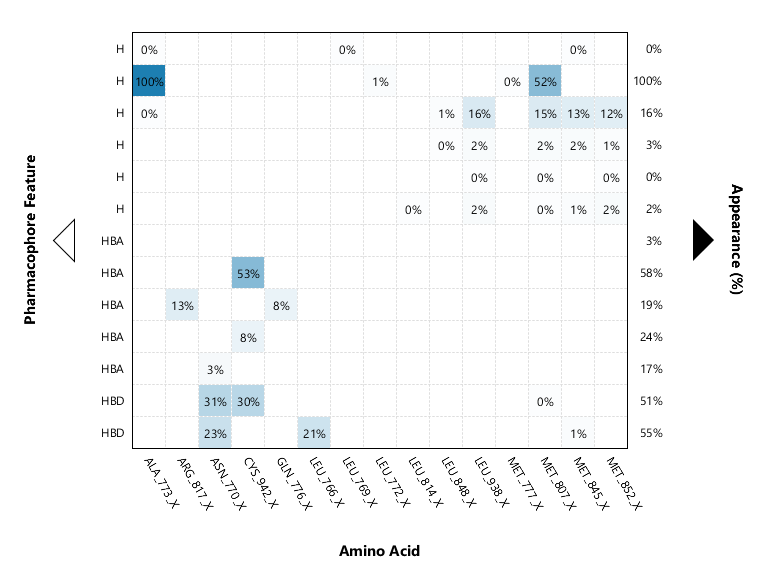
LigandScout mapas de interacción.
Farmacoforos, y residuos con mayor participación . obtencion del mapa de interaccion
Uso VMD, NAMD ENERGY
Calculo de Energias VDW, y Elec. Es necesario poseer .psf .pdb .dcd. y especificar los 2 componentes a medir. Basic […]
VMD Distance Measurements of a Simulation in NAMD
uso se carga el script, con source distance.tcl y luego Se necesita modificación en el uso de las comas/puntos para […]


(Chemistry at HARvard Macromolecular Mechanics)
CHARMM Es un programa de simulación molecular aplicable a una gran variedad de sistemas conformados por cantidades considerables de partículas, […]
2AAX with HCY vs 2AA2 with AS4 molecular dynamics
Calculos de strides optimos para los 50.000.000 frames, a los 100 ns. = 4 . Lo que da resultado 2500 […]

Procedimiento DM MR/adosterona.
http://www.charmm-gui.org/?doc=input/ligandrm Primero debemos cerciorarnos que el ligando corresponde, y no forma anillos anexos como es el caso, de AS4 dentro […]

MD 2AA2 With AS4 (Aldosterone) Production
Descargar todos los resultados https://www.dropbox.com/sh/do6utqzuajdr7as/AADcdnAnV-4WbJHGW_LUFZ90a?dl=0

RMSD analysis of trajectory (DCD file) using VMD
Mohamed shehata This video shows you how to calculate the RMSD analysis of aa protein using the software VMD from […]